第2目標:TCGA dataからNormal Tumorの情報を分割してDEGを取得する。(行動1 : tumorとnormalを分割したデータを作成する)
前回、目標1において作成したTCGA-BRCAのデータには、末尾に-01, -06,-11の3種類のデータがありそうなことが分かりました。TCGAデータには、TumorとNormalがあることが分かっているので、それぞれが何を意味しているのかGDC portal GDC にいっていくつかの情報を収集してみました。
その結果おそらく、下2桁はそれぞれ以下のサンプル種を出すことだと想定されました。
Sample Type ID : 01 => Sample Type : Primary Tumor Sample Type ID : 06 => Sample Type : Metastatic Sample Type ID : 11 => Sample Type : Solid Tissue Normal
今回、MetastaicもTumorであることを踏まえると、-01と-06をBRCA_Tumorとして、-11をBRCA_Normalとして分割したデータを作成することでTumorとNormalを分けられることが想定されました。そこで、今回はTCGA_BRCAデータをsample IDに基づいてTumorとNormalに分割することを目指したいと思います。
行動1 : tumorとnormalを分割したデータを作成する
行動1-1 : Sample typeに合わせたsample listの抽出を行う。 目標1の中で作成したTCGA_BRCAには、column namesにsample IDが入っていることを踏まえて、column namesのなかでサンプルの種類に対応する "-01", "-06", "-11"を含んでいるsample IDを抽出することが重要になります。そういった場合には、grep() functionを使用することが重要になります。そこで、3種類を以下のように減らしていきたいと思います。
BRCA_Tumor <- grep("-01", colnames(tpm1_BRCA) , value = TRUE)
BRCA_Tumor_Met <- grep("-06", colnames(tpm1_BRCA) , value = TRUE)
BRCA_Normal <- grep("-11", colnames(tpm1_BRCA) , value = TRUE)
行動1-2 : Tumorとmetastatisを合わせたsample listを作成する。 最初に記載した通り、tumorとNormalに分けるためには、TumorとTumor_metのsample listを合わせる必要があります。そこで、前回目標1-行動3で使用した積集合を求めるintersect() functionと類似の機能として和集合を求めるunion() functionを用いて、BRCA_Tumor, BRCA_Tumor_Metに含まれるsample listを作ります。
BRCA_Tumor2 <-union(BRCA_Tumor,BRCA_Tumor_Met)
行動1-3 : TumorとNormalのTPMのData tableを作成して保存する。 前回、目標1-行動3において、実施したことと同様にsample listを用いてselect() functionを用いて、作成したTumorとNormalのsample IDを保有するサンプル列を抽出してcsvファイルとして保存します。
Tpm_BRCA_Tumor <- tpm1 %>% select(sample,one_of(BRCA_Tumor2)) write.csv(Tpm_BRCA_Tumor, file = "TPM_BRCA_Tumor.csv") Tpm_BRCA_Normal <- tpm1 %>% select(sample,one_of(BRCA_Normal)) write.csv(Tpm_BRCA_Normal, file = "TPM_BRCA_Normal.csv")
ここまででTCGA-BRCA data setから、Tumor, Normalデータを抽出することができました。
今後にやっていきたいこと(随時更新予定)
今後やっていきたい目標
① TCGAに含まれる正常組織との区別をどうするか:
>目標2:行動1で解決済み
biologist-programming-training.hatenablog.com
② TPMデータを用いてDEGを算出する to adjacenyt normal tissue :
>DEGを求める手法を学習し(特性を理解し)、実際に使用する。
③ Normalに対してTumorのGSEA解析を実施する。
>GSEAを学習し(特性を理解し)、実際に使用する。
④ Tumor sampleのsub-classを探索して、性質を解析する。
④-1) Tumorの中にsubtypeが存在することを踏まえ、ばらつきの大きい遺伝子を用いてClusteirngを行う
④-2) Tumor内でのGSEA解析を実施する
⑤ single sample GSEA解析を行うことでTumorのgene set levelでの違いや特性を理解する。
上記は、1つの癌腫にこだわっての解析を記載していますが、TCGAのデータがPAN-Cancerで存在することを踏まえて、上記の解析以降は 癌腫間の比較を行いたい場合はどうするか?考えていきたいと思います。
第1目標:TCGA dataの各癌腫ごとのTPMデータの取得 (行動3:ある特定の癌腫のsubjectデータの抽出)
前回、行動2の中で今回のデータは行列に以下の情報が格納されていることが分かりました。 ・行:各geneの情報 ・列:subjectの情報
行動3 : 特定の癌種のsubjectデータの抽出(例. Breast Cancer/BRCAを抽出)
それでは、目的の癌種の情報を抽出するためには、各癌種のサンプルのみを抽出する必要があることが分かりました。 そこで、今回は、あらたに癌種の情報を取得して目的癌腫のsubjectデータの抽出を行います。
行動3-1 : 癌種情報の取得
癌腫情報を取得するために、再度UCSC xenaへ行って必要なデータを取得します。以下へaccessしてみてください。
dataset: phenotype - Curated clinical data UCSC Xena ダウンロードしたファイルを、解析用のディレクトリに格納し、read_tableもしくはfreadいずれかの方法で読み込んだのち、どういった構造のデータかview() functionで眺めてみてください。
subject1 <- read_table("TSurvival_SupplementalTable_S1_20171025_xena_sp")
view(subject1) #読み込んだtableを表示させてみる。
データを見てみると、subjectの情報(列名: sample), データセット名(列名: cancer)をはじめとして、様々なデータを含んでいることが分かりました。 →列名のCancerに基づいてsubject列を抽出することができれば癌腫に限定されたデータになることが想定されます。
行動3-2 : どの癌種情報が含まれるかの確認
先ほどview() functionを用いてデータを見たところ、3列目が各sampleの癌種情報を定義する列であることが分かりました。ただし、このままでは、どういった癌腫が含まれるのか全く分からない状況です。 そこで、列の中でuniqueなデータのみを抽出するdistinct() functionを適応して、癌腫情報のlistを取得したいと思います。(Excelを使う方はわかるかもしれませんが、Excelでいうところのデータの重複除去と同様の操作になります。)
cancer_types<-distinct(subject1[ , 3]) #distinct() functionと3列目を指定することでcancerに含まれる情報の中から重複情報のみを選択する、 view(cancer_types)
TCGA-PANCANのsubjectの情報と結びついている癌腫は以下のものがあることが分かりました。
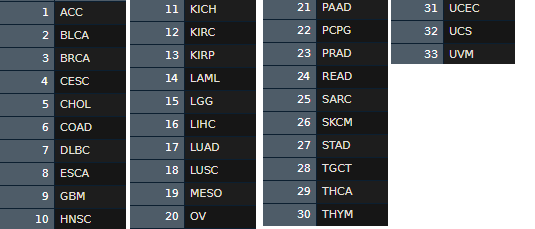
行動3-3 : BRCA(乳がん)の患者リストの作成
まずは、先ほど作成したsubject1というpatient情報のまとめから3列目の"cancer"がBRCAに該当しているsample IDのみを抽出したいと思います。 3-3-1) cancer 列が "BRCA"と一致するIDを抽出 このような場合、tidyverseに入っているfilter() functionを使います。コードは以下のようになります。
subject1_BRCA <- subject1 %>%
filter(cancer == "BRCA")
print(subject1_BRCA[1:5, 1:5],rownames=TRUE,colnames=TRUE) #出力したsubject1_BRCAを表示します。
"%>%"という初めて出てくる記号がありますが、これはtidyverseにおいて非常に多用されるパイプ演算子というもので、処理を引き続けて十する際に使用します。今回はsubject1というデータセットに対してcancerが"BRCA"という文字列の行を抽出する(フィルタリングする)という操作を行っています。これで、BRCAのサンプルIDのみを抽出することができました。 3-3-2) 乳がんsubjectのSample IDをlistとして出力する ① 今は乳がんsubjectのsample IDだけあればよいので、該当するデータを含む列(ここでは1列目を選択します。) ② 前回読み込んだtpm1もしくはtpm2(ここではtpm2を使用します。)の列名のうち、RNAseqのデータを持っているsubjectのみを選択するために列名のリストを作成します。名前の通り、colnames() というfunctionを使用します。 ③ ①(BRCAのサンプルリスト)と②(RNAseqデータがあるサンプルリスト)に共通しているsample nameのlistを、intersect() functionを用いて抽出します(=RNAseqデータを持っているBRCAサンプルのリストの作成)。イメージとしては集合の重なりを抽出する。 ④ tpmのtibbleもしくはdata.frameの中から③のlistのサンプルのデータを抽出します。
# 操作① : subject1_BRCAから1列目を抽出して"Sample_ID_BRCA"に格納 Sample_ID_BRCA <-unlist(list(subject1_BRCA[,1])) # 操作② : tpm1の列名(column names)を”Col_names_tpm1"に格納 Col_names_tpm1 <-colnames(tpm1) # tpm2の列名のlistを作成した。 # 操作③ : "Sample_ID_BRCA"と"Col_names_tpm2"に共通するsampleを"Intersect_BRCA"に格納 Intersect_BRCA <- intersect(Sample_ID_BRCA,Col_names_tpm1) # 操作④ : tpm2の中から、sample列(gene IDを含む列)と"Intersect_BRCA"の中に含まれる列名を持つデータを抽出して"tpm2_BRCA"に格納 tpm1_BRCA <- tpm1 %>% select(sample,one_of(Intersect_BRCA))
これで、一例としてBRCAのTPMデータを抽出することができました。 以降の解析で、ここからスタートできるように、csvファイルとしてwrite.csv() functionを用いて保存したいと思います。
write.csv(tpm1_BRCA, file = "TPM_BRCA.csv")
時間ができたときに、今日使用したfunctionについてのメモを作りたいと思います。
Rのデータ型とデータ構造の種類について
TCGAデータの読み込みに際して、class() functionを使用した際に、いくつかのデータ構造が出てきました。 それぞれ、どのような性質があるのかまとめておきたいと思います。
データ型
Rにおけるデータ型にはいくつかあるようですが、直近は関係ありそうなものをまとめたいと思います。
1) Character : 文字列型 "Homo Sapiens", "Mus musculus","TP53"などの文字列を表す際に使用するデータ型です。文字列は、数字であっても " で囲むことによってcharacterとして認識されます。(ex. "3", "10+2i")
2) Numeric-integer :数値-整数型 数値データのうち、整数データをintegerといいます。 (ex, 1, 100, -10)
3) Numeric-double: 数値型-実数型 数値のうち、小数点を含むデータ型をdouble といいます。 (ex. 1.045, 1e-15)
4) Logical : 論理型 TRUE, FALSE, T, Fなど今後functionとの組み合わせなどで使用する論理を示すデータ型です。
データ構造
データ型を格納するデータ構造の種類の一部をまとめたいと思います。
1) Vector(ベクター)型 同じデータ型のデータを格納する。同じというところが肝で、数値なら数値で構成されたベクター、文字列なら文字列のベクターということになります。
numeric_vector <- c(1, 4, 3, 5) # 整数型ベクター
character_vector <- c ("TP53", "RB", "CDK6") # 文字列型ベクター
2) Matrix(行列)型 行列は高校の数学で習ったことを覚えているかもしれませんが、行と列で構成されたデータ構造になります。行列の要素はNAや数値データなどが入ることになります。
Matrix_1 <- matrix(c(1, 4, 3, 5), nrow = 2) #行列の作成方法
nrowで行数を設定して行数を設定するので、ここでは2x2の行列が出来上がります。
3) data.frame型 行列と似た構造をしていますが、数値だけでなく、異なるデータ型を1つにまとめることができます。 また、行名と列名の両方を持つことできるというのは4)のtibble型との違いになります。
4) tibble型 tidyverseというRでよく使われるpackage群にて、data.frameの代わりに使用されるデータ構造になります。tidyverseのなかのreadr packageに含まれるread_table()を使用した際に出力されたデータ構造かと思います。
・データの性質を理解しやすい。 ・どのようなデータ型のデータも格納できる。 ・tibble型は行名を許容しない (これがTCGAデータの解析には重要な気がします)
Reference.
第1目標:TCGA dataの各癌腫ごとのTPMデータの取得 (行動2:ディレクトリの設定、packageのinstallと呼び出し、データの読み込みと構造の理解)
それでは、ここからはダウロードしたデータ ("tcga_RSEM_gene_tpm.gz")を使用して、ある癌腫のデータの抽出に進みたいと思いますが、今日はファイルの格納場所を設定し、ファイルを読み込む方法を記載したいと思います。
行動2 データを読み込み、構造を理解する。
2-1) データ格納フォルダをRに教える (ディレクトリの設定)
以前(R Studioの設定)にて右下にある"Environment,History~"の中で、"File"タブをクリックすると、現在defaultで選択されている作業ディレクトリが表示されます。
TCGAデータが格納されている場合(もしくは、ファイルを表示ディレクトリに格納している場合)はそのままでよいのですが、筆者は今後のために解析用に新しいフォルダ"Blog-TCGA-PANCAN"を作成し、前回downloadした2ファイルを格納したので、そこを設定する方法を表示します。
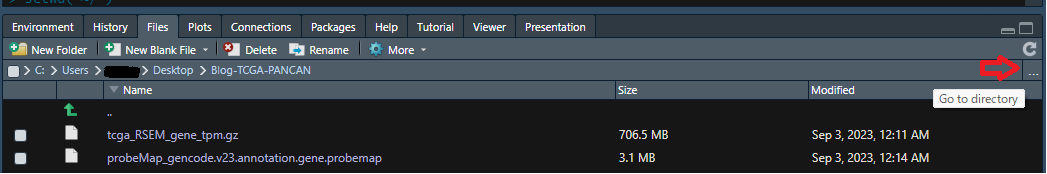
2-1-1) 赤矢印の”...”をクリックするとdirectoryを選択するためのwindowが開くので、そこで該当するフォルダ(ここでは”Blog-TCGA-PANCAN”)を選択します。
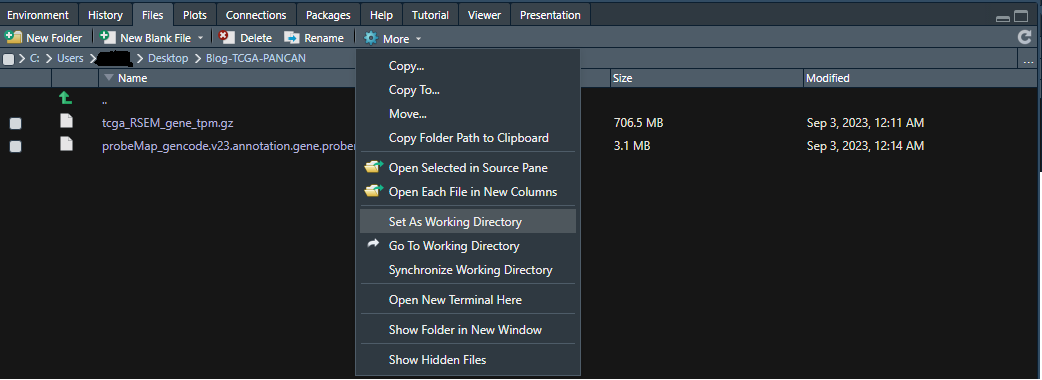
2-1-2) 操作1ではフォルダを開いただけで設定はできていないので、次に⚙(歯車アイコン)Moreをクリックすると以下のような選択肢が現れるので、"Set As Working Directory"を選択してください。これで設定完了。
実際には "setwd()" というfunctionを用いたのと同じ状況になります。
念のため、自分のやり方でよいのか不安になり調べると、ネットには非常に多くの情報がありました(Ref.1)。先駆者様様です。念のためご紹介しておきます。
2-1 別法) Toolsタブ > Global Options... > Generalメニューの中で、”Default working directory(when not in a project)”のBrowseボタンを押して該当フォルダを選択する。
Reference.1作業ディレクトリの設定について
mom-neuroscience.com
mom-neuroscience.com
2-2 解析に必要なpackageをinstallして、パッケージを呼び出す
今回の目標を達成するためには、 ダウンロードしたTCGAデータ(tcga_RSEM_gene_tpm.gz)を読み込む必要があります。
2-2-1) 必要なpackageのインストール
そこで、Rで解析を行うためのパッケージ(package, 解析機能の詰め合わせみたいなもの)をインストールして、R-studio上で使えるようにする(=呼び出す)必要があります。
packageのインストール方法は複数ありそうですが、2つを説明します。
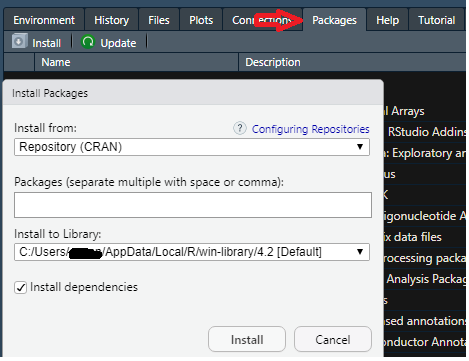
2-2-1-1)R Studioの右下のPackages Tab(赤矢印のtab)を開き、インストールしたいパッケージを入力してダウンロードします。
2-2-1-2) install.packagesを利用してinstallする方法があります。
install.packages(パッケージ名)
2-2-2) 必要なpackageの呼び出し
library functionを用いて、呼び出すpackageを指定します。
library(パッケージ名)
2-3 TCGA dataを読み込む
今回のTCGAデータの読み込む方法としてはいくつかの方法がありそうです。
1) Rに標準で設定されているread.table()を使用する。
tpm1 <- read.table("tcga_RSEM_gene_tpm.gz") #tpm1 という変数にtcga dataを格納する。
class(tpm1) # 格納されたtpm1 のデータtypeを確認する=>"spec_tbl_df" "tbl_df""tbl""data.frame" 型として呼び込まれた。
1) readrもしくはtidyverseというpackageに入っているread_table()を使用する。
install.packages(tidyverse) #tidyverse packageをinstallする
library(tidyverse) #tidyverse packageを呼び出す
tpm1 <- read_table("tcga_RSEM_gene_tpm.gz") #tpm1 という変数にtcga dataを格納する。
class(tpm1) # 格納されたtpm1 のデータtypeを確認する
print(tpm1[1:5, 1:5],rownames=TRUE,colnames=TRUE) # 1-5行目 x 1-5列目までを表示=>"tbl"(tibble)型として呼び込まれた。
2) data.tableというpackageに入っているfread()を使用する。
install.packages(data.table) #data.table packageをinstallする
library(data.table) #data.table packageを呼び出す
tpm2 <- fread("tcga_RSEM_gene_tpm.gz") #tpm2 という変数にtcga dataを格納する。
class(tpm2) # 格納されたtpm1 のデータtypeを確認する
print(tpm2[1:5, 1:5],rownames=TRUE,colnames=TRUE) # 1-5行目 x 1-5列目までを表示=> "data.table" "data.frame"型として呼び込まれた。
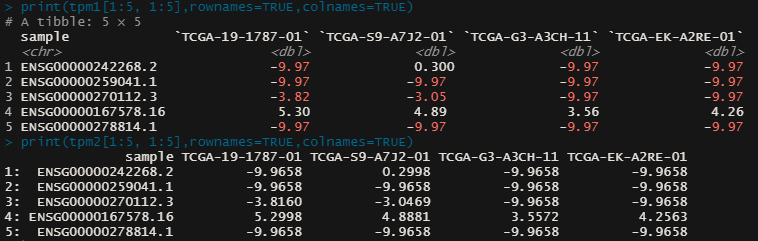
データを見ていただければわかる通り今のtibble, data.frameは1行目に各geneに該当するEnsemble IDの情報が、列方向に各サンプルの情報が並んでいることが分かりました。
tibble, data.frameの違いの説明を含むRのデータ型・データ構造については以下を確認ください。
biologist-programming-training.hatenablog.com
間話 : 今後解析に使用するPCの性能とRのversion情報 (参考URL含む)
今後、解析を進めていくうえで新たに購入したPCがどこで時間がかかるのかを含めて記録するためにPCの性能、Rプログラムのversion情報などをメモしていこうと思います。が、何の情報を書いたらよいかわからないので、とりあえず、
メーカー : hp
モデル :HP Pro SFF 400 G9
プロセッサ :12th Gen Intel(R) Core(TM) i5-12500 3.00 GHz
RAM :32.0GB
今は、NGSの解析パイプラインなど組むことは全く想定していないのでとりあえず、32GBにしてみました。もし、解析に不足を感じ、お財布に余裕ができたらメモリを乗せ換えることを検討したいと思います。
また、基本は唯一筆者が少しだけかじっているRをベースに解析を進めていこうと思います。データ解析に対してPythonのほうが良いような場面も見るので、いつか気が向いたら(何年後になるかわかりませんが、、、)Pythonでの解析も考えたいと思います。
R : version 4.2.2
R-Studio : 2022.07.2 Build 576
RとR studioのダウンロードの仕方はいろいろなところで書いてあるので以下に自分が参考にさせていただいたURLを張っておきます。
参考 1) 東大 門田先生のアグリバイオインフォマティクスのページ
参考 2)宋先生と 矢内先生が共同で執筆するRプログラミングの「入門書」
下準備 : R-studioの画面設定
本格的に解析に入る前に、R-studioの設定をしたいと思います。インストール後とりあえず、以下のようにして操作画面のPanel設定をします。
1) "Tools"タブの”Global Options...”を押してメニューを開きます。
2) Appearanceを選択し、自分の好みに合う"Editor font:"と"Editor theme"を選択します。
(筆者は、画面が黒そうなほうが目によさそう & かっこいいという理由からfont:Lucida Consoleとtheme : Chaosとしています。)
3) Panel Layoutを設定する。
(筆者設定はRの勉強中にどこかで見た設定を参考に、以下のように設定しています。)
左上 : Source
右上 : Console
左下:未選択 (Sourceを縦長に展開するため)
右下:Environment, History, Files, Plot~を選び、すべてにCheckを入れる。
まぁ以下のような感じです。
では、次以降進めていきましょう。
第1目標:TCGA dataの各癌腫ごとのTPMデータの取得 (行動1:TCGAデータの取得)
まず、1つ目の目標として、がん領域において非常に重要なTCGAデータのダウンロードと、その中身の確認方法を検討していきたいと思います。
それでは、1つめの目標をまずタイトルの通りに「TCGA dataの各癌腫ごとのTPMデータの取得」と設定することにします。今回はまず、TPMデータのダウンロードまでを書いていきたいと思います。
1. UCSC Xenaにアクセスする。(https://xena.ucsc.edu/)
1-1 : "Launch Xena"ボタンを押して次に進む。
このあたりの詳細は統合TV(https://togotv.dbcls.jp/en/20200313.html)でも動画あります。自分が目指すのは、Rawデータにより近いところからの解析なので、Visuzalizationのパートは統合TVを見ればよいかと思います。
1-2 : ページの左上にある"DATA SET" タブを押して次に進む。
これによって、膨大なデータが選択できるようになるはずです。執筆時で129 Cohorts, 1571 Datasetsという感じでした。
1-3 : TCGA Pan-Cancer (PANCAN)を選択する。
同じようなデータに対して各癌腫間の比較が将来の目標なので、データ量は膨大ですが、”cohort: TCGA Pan-Cancer (PANCAN)”を選択してみたいと思います。
1-4 : gene expression RNAseqのTOIL RSEM tpm (n=10,535) UCSC Toil RNA-seq Recomputeを開く
TCGAってすごいと毎回感服させられますが、いろいろなデータが格納されているようです。(一応、この時点のURLも張っておこうと思います。https://xenabrowser.net/datapages/?cohort=TCGA%20Pan-Cancer%20(PANCAN))
ここで、問題発生です。
第一目標はTPM情報の取得なのですが、いろいろなデータがありそうですが、TPMとして想定されるデータとしては以下の2つが見つかりました。
① gene expression RNAseqのTOIL RSEM tpm (n=10,535) UCSC Toil RNA-seq Recompute
② transcript expression RNAseqのTOIL RSEM tpm (n=10,535) UCSC Toil RNA-seq Recompute
後半のファイル名が同じ?!という感じですが、両URLを開いてみるとURLにもそれぞれ"RSEM_gene_tpm"と"rsem_isoform_tpm"とそれぞれ記載があり、とんだ先のページの説明のTableにおいても、それぞれのidentifierが
・60,499 identifiers X 10535 samples
・198,620 identifiers X 10535 samples
と全く異なっておりました、URLの名づけを踏まえても、後者はGeneのisoformごとに分割したファイルという感じかなと(勝手に)理解し、突き進むことにしました。
(もし違っていたらいろいろと終りますが、、、、まぁそこは初心者ということで容赦いただくことにしましょう。)
1-5 : downloadの横にある"~/tcga_RSEM_gene_tpm.gz"というファイルと、ID/Gene Mappingの横にある"~/probeMap%2Fgencode.v23.annotation.gene.probemap;"というファイルをダウンロードする。
1-4の説明でidentifiersとサンプル数の数を見た人は薄々感じているかもしれませんが、1つめのTCGA PANCANの約1万例x6万遺伝子?(coding以外も入っているってこと??)のファイルは、結構重めの723Mbでした。
2つ目のファイルは、ファイル名をみればそのままで、今のidentifierとgene nameをつなげるために必要なファイルであろうと想定されます。こちらはあまり大きなファイルではないですね。
とりあえず、第1目標の行動1としてここまでにしたいと思います。