第1目標:TCGA dataの各癌腫ごとのTPMデータの取得 (行動2:ディレクトリの設定、packageのinstallと呼び出し、データの読み込みと構造の理解)
それでは、ここからはダウロードしたデータ ("tcga_RSEM_gene_tpm.gz")を使用して、ある癌腫のデータの抽出に進みたいと思いますが、今日はファイルの格納場所を設定し、ファイルを読み込む方法を記載したいと思います。
行動2 データを読み込み、構造を理解する。
2-1) データ格納フォルダをRに教える (ディレクトリの設定)
以前(R Studioの設定)にて右下にある"Environment,History~"の中で、"File"タブをクリックすると、現在defaultで選択されている作業ディレクトリが表示されます。
TCGAデータが格納されている場合(もしくは、ファイルを表示ディレクトリに格納している場合)はそのままでよいのですが、筆者は今後のために解析用に新しいフォルダ"Blog-TCGA-PANCAN"を作成し、前回downloadした2ファイルを格納したので、そこを設定する方法を表示します。
2-1-1) 赤矢印の”...”をクリックするとdirectoryを選択するためのwindowが開くので、そこで該当するフォルダ(ここでは”Blog-TCGA-PANCAN”)を選択します。
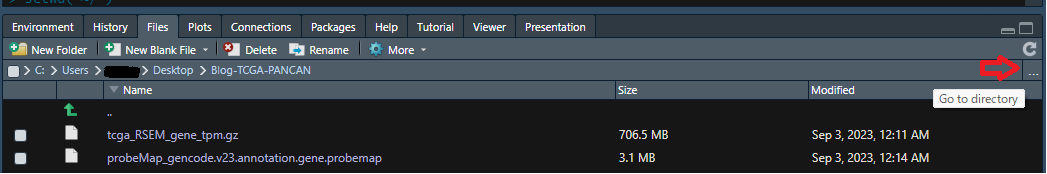
2-1-2) 操作1ではフォルダを開いただけで設定はできていないので、次に⚙(歯車アイコン)Moreをクリックすると以下のような選択肢が現れるので、"Set As Working Directory"を選択してください。これで設定完了。
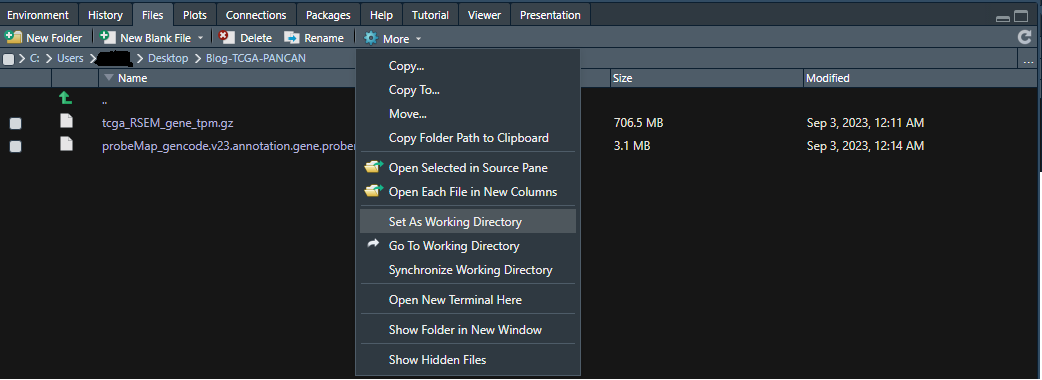
実際には "setwd()" というfunctionを用いたのと同じ状況になります。
念のため、自分のやり方でよいのか不安になり調べると、ネットには非常に多くの情報がありました(Ref.1)。先駆者様様です。念のためご紹介しておきます。
2-1 別法) Toolsタブ > Global Options... > Generalメニューの中で、”Default working directory(when not in a project)”のBrowseボタンを押して該当フォルダを選択する。
Reference.1作業ディレクトリの設定について
mom-neuroscience.com
mom-neuroscience.com
2-2 解析に必要なpackageをinstallして、パッケージを呼び出す
今回の目標を達成するためには、 ダウンロードしたTCGAデータ(tcga_RSEM_gene_tpm.gz)を読み込む必要があります。
2-2-1) 必要なpackageのインストール
そこで、Rで解析を行うためのパッケージ(package, 解析機能の詰め合わせみたいなもの)をインストールして、R-studio上で使えるようにする(=呼び出す)必要があります。
packageのインストール方法は複数ありそうですが、2つを説明します。
2-2-1-1)R Studioの右下のPackages Tab(赤矢印のtab)を開き、インストールしたいパッケージを入力してダウンロードします。
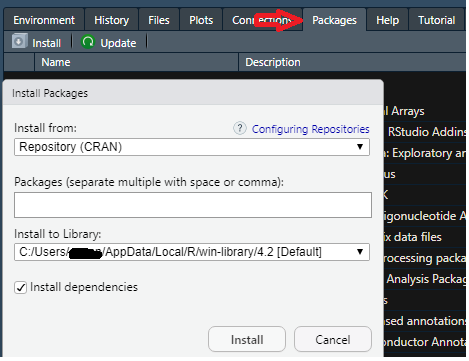
2-2-1-2) install.packagesを利用してinstallする方法があります。
install.packages(パッケージ名)
2-2-2) 必要なpackageの呼び出し
library functionを用いて、呼び出すpackageを指定します。
library(パッケージ名)
2-3 TCGA dataを読み込む
今回のTCGAデータの読み込む方法としてはいくつかの方法がありそうです。
1) Rに標準で設定されているread.table()を使用する。
tpm1 <- read.table("tcga_RSEM_gene_tpm.gz") #tpm1 という変数にtcga dataを格納する。
class(tpm1) # 格納されたtpm1 のデータtypeを確認する=>"spec_tbl_df" "tbl_df""tbl""data.frame" 型として呼び込まれた。
1) readrもしくはtidyverseというpackageに入っているread_table()を使用する。
install.packages(tidyverse) #tidyverse packageをinstallする
library(tidyverse) #tidyverse packageを呼び出す
tpm1 <- read_table("tcga_RSEM_gene_tpm.gz") #tpm1 という変数にtcga dataを格納する。
class(tpm1) # 格納されたtpm1 のデータtypeを確認する
print(tpm1[1:5, 1:5],rownames=TRUE,colnames=TRUE) # 1-5行目 x 1-5列目までを表示=>"tbl"(tibble)型として呼び込まれた。
2) data.tableというpackageに入っているfread()を使用する。
install.packages(data.table) #data.table packageをinstallする
library(data.table) #data.table packageを呼び出す
tpm2 <- fread("tcga_RSEM_gene_tpm.gz") #tpm2 という変数にtcga dataを格納する。
class(tpm2) # 格納されたtpm1 のデータtypeを確認する
print(tpm2[1:5, 1:5],rownames=TRUE,colnames=TRUE) # 1-5行目 x 1-5列目までを表示=> "data.table" "data.frame"型として呼び込まれた。
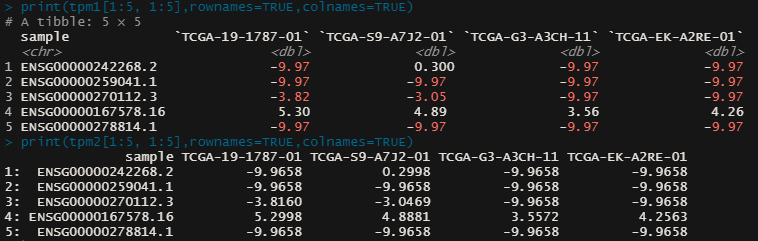
データを見ていただければわかる通り今のtibble, data.frameは1行目に各geneに該当するEnsemble IDの情報が、列方向に各サンプルの情報が並んでいることが分かりました。
tibble, data.frameの違いの説明を含むRのデータ型・データ構造については以下を確認ください。
biologist-programming-training.hatenablog.com